These are turbulent times for the Food and Drug Administration. The almost daily barrage of headlines questioning the safety of marketed drugs is probably depleting regulators' personal stocks of aspirin and antacids. But as they try to soothe their own pain, regulators must not forget their mission: to ease the plight of patients who need new medicines.
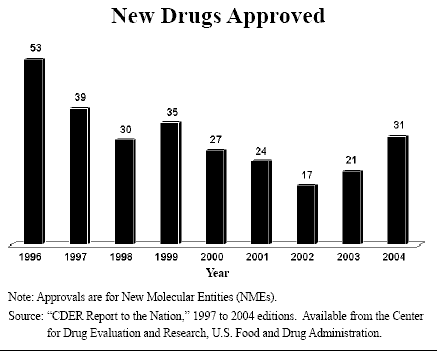
Risk vs. Reward. Past criticism of the FDA centered mostly on what arguably remains the agency's most important shortcoming: the delay and escalating expense of getting drugs through the development pipeline and into the market. The development of drugs in the United States is in real trouble:
- Costs to bring an average drug to market have skyrocketed and now exceed $800 million.
- Fewer than one-in-three approved drugs ever recoup their development costs.
- Ominously, the number of applications to the FDA to approve the marketing of new drugs has decreased steadily since 1995.
But since drug and biotech companies spend more than $30 billion a year on research, how can that be? The reason is that regulators are continually raising the bar for approval – and the number of drugs approved by the FDA each year has fallen [see the figure].
FDA reviewers are conditioned to be risk-averse: As former FDA General Counsel Peter Barton Hutt has observed, "FDA employees have been praised only for refusing to approve a new drug, not for making a courageous judgment to approve a new drug that has in fact helped patients and advanced the public health."
As a result, regulators, nearly phobic about risks, make decisions defensively, at the cost of denying patients drugs that can cure diseases and save lives. They tend to delay or reject new products of all sorts, from fat substitutes to vaccines to painkillers. That's bad for public health and for consumers' freedom to choose.
Erring on the Side of Safety. When the FDA grants marketing approval, the drug is deemed safe and effective for conditions on the label. However, the "safety" of a drug is relative. Regulators' judgments require a global and often difficult calculation of risk and benefit, including consideration of available alternatives. For example, we tolerate more uncertainty and severe side effects from a potential cure for pancreatic cancer or AIDS than for treating heartburn.
Lately, however, events have conspired to increase even further the pressure on regulators to err on the side of "safety." First there were claims that the labels of certain antidepressants, such as Paxil, failed to warn that the drugs caused some adolescents to commit suicide. Then the agency was blind-sided by contamination that rendered unusable half the nation's flu vaccine last winter. Finally, there were revelations about previously unknown side effects of several widely prescribed anti-inflammatory analgesic drugs – so-called Cox-2 inhibitors – which appear to increase the risks of heart attack and stroke.
Case Study: Drugs to Treat Multiple Sclerosis. Regulators' increasing sensitivity to safety may have become contagious: Drug manufacturers, too, seem to have begun to "err on the side of safety," and have voluntarily removed safe and effective drugs from the market. Consider Tysabri, only the sixth medication approved – and the first in several years – to treat multiple sclerosis (MS), a debilitating autoimmune disease affecting the central nervous system. The results of clinical trials were stunning: the frequency of clinical relapses was cut by more than half. This induced the FDA to grant accelerated approval in late 2004. MS patients eagerly put their names on waiting lists for the medicine.
But this ray of hope for MS sufferers was short-lived. By the time several thousand patients were being treated with Tysabri, three confirmed cases of a rare neurological disorder (progressive multifocal leukoencephalopathy, or PML) caused by a virus were reported. (Because the drug suppresses the immune response, regulators, clinicians and the drug's developers were sensitive from the beginning to infections as a possible side effect.)
Immediately – some say prematurely – the medicine's manufacturers voluntarily halted production and distribution and withdrew Tysabri from the market. MS victims and many neurologists were bitterly disappointed. Now they can only hope a comprehensive review of all the clinical data – which confirms the drug's efficacy and has revealed no new cases of PML – will permit the drug's return to the market, perhaps with new labeling.
Other Case Studies. Another example of a drug's hasty withdrawal is Lotronex, which is used to treat irritable bowel syndrome. About one in every 350 women who took it suffered ischemic colitis – a serious side effect in which intestinal blood flow is compromised. However, the drug posed a relatively small risk to the other 349, who had no other effective treatment available. In 2002, after thousands of patients lobbied the FDA for its return, Lotronex was returned to the market.
Patient Choice. As more breakthrough drugs come before the FDA for approval, the agency must find a way to balance safety with the monumental costs of drug testing and patients' right to assume responsibility for their own medical decisions. This would be a sea change for the FDA, which has sought repeatedly to limit physicians' and patients' discretion on treatment decisions, including restricting physicians from prescribing drugs for conditions not listed on the labels ("off-label" use), and interfering with drug companies' ability to disseminate new information about drug therapy.
After consulting with health care professionals, patients should be able to make informed decisions about possible treatment options. That fundamental right should not be usurped by risk-averse, publicity-shy bureaucrats, anti-FDA health care activists or members of Congress.
The notion that the FDA should "err on the side of safety" must be qualified for patients with incurable or poorly treatable diseases, as in the cases of Tysabri and Lotronex discussed above. For those patients, there is no safety in the status quo. If the FDA must err, it should be on the side of patients' freedom to choose.
Conclusion. We need to insulate policy-making and decisions on individual products from politics to the extent possible, require regulators to make decisions based on the facts and improve pharmacovigilance – the safety monitoring of drugs already on the market. Finally, and most important, we need to reform the culture of excessive risk-aversion and defensive decision-making that pervades the FDA.
Henry I. Miller, M.D., is an NCPA senior fellow, a research fellow at the Hoover Institution and a former Food and Drug Administration official.