- The major causes of cancer are:
a) Smoking: About a third of U.S. cancer (90 percent of lung cancer);
b) Dietary imbalances, e.g., lack of dietary fruits and vegetables: The quarter of the population eating the least fruits and vegetables has double the cancer rate for most types of cancer compared to the quarter eating the most;
c) Chronic infections: mostly in developing countries; and
d) Hormonal factors: primarily influenced by life style. - There is no epidemic of cancer, except for lung cancer due to smoking. Cancer mortality rates have declined 16 percent since 1950 (excluding lung cancer).
- Regulatory policy that focuses on traces of synthetic chemicals is based on misconceptions about animal cancer tests. Recent research indicates that:
a) Rodent carcinogens are not rare. Half of all chemicals tested in standard high dose animal cancer tests, whether occurring naturally or produced synthetically, are "carcinogens";
b) There are high-dose effects in rodent cancer tests that are not relevant to low-dose human exposures and that contribute to the high proportion of chemicals that test positive;
c) The focus of regulatory policy is on synthetic chemicals, although 99.9 percent of the chemicals humans ingest are natural. More than 1,000 chemicals have been described in coffee: 28 have been tested and 19 are rodent carcinogens. Plants in the human diet contain thousands of natural pesticides that protect them from insects and other predators: 63 have been tested and 35 are rodent carcinogens. - There is no convincing evidence that synthetic chemical pollutants are important for human cancer. Regulations that try to eliminate minuscule levels of synthetic chemicals are enormously expensive: The Environmental Protection Agency has estimated that environmental regulations cost society $140 billion per year. Others have estimated that the median toxic control program costs 146 times more per life year saved than the median medical intervention. Attempting to reduce tiny hypothetical risks also has costs; for example, if reducing synthetic pesticides makes fruits and vegetables more expensive, thereby decreasing consumption, then cancer will be increased, particularly for the poor.
- Prevention of cancer will come from knowledge obtained from biomedical research, education of the public and lifestyle changes by individuals. A re-examination of priorities in cancer prevention, both public and private, seems called for.
Various misconceptions about the relationship between environmental pollution and human disease, particularly cancer, drive regulatory policy. In this paper, we highlight 10 such misconceptions and briefly present the scientific evidence that undermines each.
[page]Cancer death rates overall in the U.S. (excluding lung cancer due to smoking) have declined 16 percent since 1950.1 If lung cancer is included, mortality rates have increased over time, but recently have declined in men due to decreased smoking.
- The types of cancer deaths that have decreased are primarily stomach, cervical, uterine and colorectal.
- The types that have increased are primarily lung cancer (90 percent is due to smoking, as are 35 percent of all cancer deaths in the U.S.), melanoma (probably due to sunburns) and non-Hodgkin's lymphoma.
"Cancer death rates overall (excluding lung cancer) have declined 16 percent since 1950."
The rise in incidence rates in older age groups for some cancers, e.g., prostate, can be explained by known factors such as improved screening.2 As one study noted, "The reason for not focusing on the reported incidence of cancer is that the scope and precision of diagnostic information, practices in screening and early detection, and criteria for reporting cancer have changed so much over time that trends in incidence are not reliable."3 Life expectancy has continued to rise since 1950.
[page]Neither the study of patterns of disease in humans (epidemiology) nor experimental studies on laboratory animals (toxicology) support this idea.4 Epidemiological studies have identified the factors that are likely to have a major effect on lowering rates of cancer: reduction of smoking, improving diet (e.g., increased consumption of fruits and vegetables), hormonal factors and control of infections. Although some epidemiologic studies find an association between cancer and low levels of industrial pollutants, the associations are usually weak, the results are usually conflicting and the studies do not correct for potentially large confounding factors like diet. Moreover, exposures to synthetic pollutants are tiny and rarely seem toxicologically plausible as a causal factor, particularly when compared to the background of natural chemicals that are rodent carcinogens.5
"The proportion of cancer that regulation could prevent would be tiny."
Even assuming that worst-case risk estimates for synthetic pollutants are true risks, the proportion of cancer that the Environmental Protection Agency (EPA) could prevent by regulation would be tiny.6 Occupational exposures to some carcinogens cause cancer, though how much has been a controversial issue: a few percent seems a reasonable estimate,7 much of this from asbestos in smokers. Exposures to substances in the workplace can be high in comparison with other chemical exposures in food, air or water. Past occupational exposures have sometimes been high and therefore comparatively little quantitative extrapolation may be required for risk assessment from high-dose rodent tests to high-dose occupational exposures. Since occupational cancer is concentrated among small groups exposed at high levels, there is an opportunity to control or eliminate risks once they are identified; however, current permitted workplace exposures are sometimes close to the carcinogenic dose in rodents.8
Cancer is due in part to normal aging and increases exponentially with age in both rodents and humans.9 To the extent that the major external risk factors for cancer are diminished, cancer will occur at a later age, and the proportion of cancer caused by normal metabolic processes will increase. Aging and its degenerative diseases appear to be due in good part to oxidative damage to DNA and other macromolecules.10 Oxidant by-products of normal metabolism – superoxide, hydrogen peroxide and hydroxyl radical – are the same mutagens (agents that alter DNA) produced by radiation. Mitochondria from old animals leak oxidants;11 old rats have about 66,000 oxidative DNA lesions per cell.12 DNA is oxidized in normal metabolism because antioxidant defenses, though numerous, are not perfect. Antioxidant defenses against oxidative damage include Vitamins C and E and perhaps carotenoids,13 most of which come from dietary fruits and vegetables.
"Smoking contributes to about 35 percent of cancer."
Smoking contributes to about 35 percent of U.S. cancer, about one-quarter of heart disease and about 400,000 premature deaths per year in the United States.14 Tobacco is a known cause of cancer of the lung, bladder, mouth, pharynx, pancreas, stomach, larynx, esophagus and possibly colon. Tobacco causes even more deaths by diseases other than cancer. Smoke contains a wide variety of mutagens and rodent carcinogens. Smoking is also a severe oxidative stress and causes inflammation in the lung. The oxidants in cigarette smoke – mainly nitrogen oxides – deplete the body's antioxidants. Thus, smokers must ingest two to three times more Vitamin C than non-smokers to achieve the same level in blood, but they rarely do. Inadequate concentration of Vitamin C in plasma is more common among the poor and smokers.
Men with inadequate diets or who smoke may damage the DNA of their sperm as well as the DNA in the rest of their cells. When the level of dietary Vitamin C is insufficient to keep seminal fluid Vitamin C at an adequate level, the oxidative lesions in sperm DNA are increased 250 percent.15 Male smokers, compared to non-smokers, have more oxidative lesions in sperm DNA16 and more chromosomal abnormalities in sperm.17 Smoking by fathers, therefore, may plausibly increase the risk of birth defects and childhood cancer in offspring.18 A new epidemiological study suggests that the rate of childhood cancers is increased in offspring of male smokers, e.g., acute lymphocytic leukemia, lymphoma, and brain tumors are increased three to four times.19
"Unbalanced diets, with a low intake of fruits and vegetables, account for about one-third of cancer risk."
The authors estimate that unbalanced diets (e.g., low intake of fruits and vegetables) account for about one-third of cancer risk, in agreement with the earlier estimate of researchers R. Doll and R. Peto.20 [See Misconception #3.] There has been considerable interest in calories (and dietary fat) as a risk factor for cancer, in part because caloric restriction markedly lowers the cancer rate and increases life span in rodents.21
Chronic inflammation from chronic infection, a major contributor to cancer,22 results in release from white cells of oxidants that are mutagens. White cells and other phagocytic cells of the immune system combat bacteria, parasites and virus-infected cells by destroying them with potent, mutagenic oxidizing agents. The oxidants protect humans from immediate death from infection, but they also cause oxidative damage to DNA, chronic cell killing with compensatory cell division, and mutation23 and thus contribute to the carcinogenic process. Antioxidants appear to inhibit some of the pathology of chronic inflammation. Chronic infections cause about 21 percent of new cancer cases in developing countries and 9 percent in developed countries.24
Reproductive hormones play a large role in cancer, including breast, prostate, ovary and endometrium (the inner lining of the uterus),25 contributing to as much as 20 percent of all cancer. Many lifestyle factors such as reproductive history, lack of exercise, obesity and alcohol intake influence hormone levels and therefore increase risk.26
Other causal factors in human cancer are excessive alcohol consumption, excessive sun exposure, and viruses. Genetic factors also play a significant role and interact with lifestyle and other risk factors. Biomedical research is uncovering important genetic variation in humans.
[page]"If fruits and vegetables become more expensive because of reduced synthetic pesticides then cancer, especially among the poor, is likely to increase."
On the contrary, fruits and vegetables are of major importance for reducing cancer; if they become more expensive because of reduced use of synthetic pesticides, then cancer is likely to increase. People with low incomes eat fewer fruits and vegetables and spend a higher percentage of their income on food.
High consumption of fruits and vegetables is associated with a lowered risk of degenerative diseases including cancer, cardiovascular disease, cataracts and brain dysfunction.27
- Over 200 studies in the epidemiological literature have been reviewed that show, with great consistency, an association between low consumption of fruits and vegetables and cancer incidence.28 [See Appendix Table I].
- The quarter of the population with the lowest dietary intake of fruits and vegetables has roughly twice the cancer rate of the quarter with the highest intake for most types of cancer (lung, larynx, oral cavity, esophagus, stomach, colorectal, bladder, pancreas, cervix and ovary).
- 80 percent of U.S. children and adolescents29 and 68 percent of adults did not meet the intake recommended by the National Cancer Institute and the National Research Council: five servings of fruits and vegetables per day.
Publicity about hundreds of minor hypothetical risks, such as pesticide residues (see Misconception #7), can cause loss of perspective on what is important: half the public does not know that fruit and vegetable consumption is a major protection against cancer.30
[page]Hormonal factors, dietary imbalances, infection and inflammation and genetic factors, none of which involve a carcinogenic chemical, are major contributors to cancer.
Deficiency of micronutrients, many of which come from fruits and vegetables, can cause cancer. Antioxidants may account for some of the beneficial effect of fruits and vegetables, as discussed in Misconception #2. However, the effects of deficiency of dietary antioxidants are difficult to disentangle by epidemiological studies from that of other important vitamins and ingredients present in fruits and vegetables.31
Folate deficiency, one of the most common vitamin deficiencies, causes extensive chromosome breaks in human genes.32 Approximately 10 percent of the US population33 has a lower folate level than that at which chromosome breaks occur.34 In two small studies of low income (mainly African-American) elderly35 and adolescents36 nearly half had folate levels that low. The rate of chromosome breaks in humans is reduced by folate administration.37 Chromosome breaks could contribute to the increased risk of cancer and cognitive defects associated with folate deficiency in humans. 38 Folate deficiency also damages human sperm,39 causes neural tube defects in the fetus and about 10 percent of U.S. heart disease.40 Diets deficient in fruits and vegetables are commonly low in folate, antioxidants, (e.g., vitamin C) and many other micronutrients, and result in DNA damage and higher cancer rates.41
Many other micronutrients whose main dietary sources are other than fruits and vegetables, also are likely to play a significant role in the prevention and repair of DNA damage, and thus are important to the maintenance of long term health. Deficiency of vitamin B12 (found in 5 percent of the U.S. population) causes a functional folate deficiency, accumulation of homocysteine (a risk factor for heart disease)42 and probably causes chromosome breaks.43 Strict vegetarians are at increased risk of developing a vitamin B12 deficiency.44 Niacin contributes to the repair of DNA breaks.45 As a result, dietary insufficiencies of niacin (15 percent of some populations are deficient)46, folate and antioxidants may act together to increase DNA damage. There is also some evidence that deficiencies of zinc, copper, iron, selenium and vitamin B6 (each deficiency occurs in 5 to 20 percent of the population), can lead to DNA damage.47 Optimizing micronutrient intake (through better diets, fortification of foods or multivitamin-mineral pills) can have a major impact on health at low cost. Increasing research in this area, and efforts to increase micronutrient intake and more balanced diets, should be high priorities for public policy.
[page]"The amounts of synthetic pesticide residues in plant foods are insignificant compared to the amount of natural pesticides produced by plants themselves."
On the contrary, 99.9 percent of the chemicals humans ingest are natural. The amounts of synthetic pesticide residues in plant foods are insignificant compared to the amount of natural pesticides produced by plants themselves.48 Of all dietary pesticides that humans eat, 99.99 percent are natural: they are chemicals produced by plants to defend themselves against fungi, insects, and other animal predators.49 Each plant produces a different array of such chemicals.
- On average Americans ingest roughly 5,000 to 10,000 different natural pesticides and their breakdown products.
- Americans eat about 1,500 mg of natural pesticides per person per day, which is about 10,000 times more than they consume of synthetic pesticide residues.
Even though only a small proportion of natural pesticides have been tested for carcinogenicity, 35 of the 63 tested are rodent carcinogens. Naturally occurring pesticides that are rodent carcinogens are ubiquitous in fruits, vegetables, herbs, and spices.50 [See Appendix Table II.]
"In a single cup of coffee the natural chemicals that are known rodent carcinogens are about equal in weight to a year's worth of synthetic pesticide residues."
Cooking foods produces about 2,000 mg per person per day of burnt material that contains many rodent carcinogens and many mutagens. By contrast, the residues of 200 synthetic chemicals measured by FDA, including the synthetic pesticides thought to be of greatest importance, average only about 0.09 mg per person per day.51 In a single cup of coffee the natural chemicals that are known rodent carcinogens are about equal in weight to a year's worth of synthetic pesticide residues that are rodent carcinogens, even though only 3 percent of the natural chemicals in roasted coffee have been adequately tested for carcinogenicity.52 [See Appendix Table III.] This does not mean that coffee or natural pesticides are dangerous, but rather that assumptions about high dose animal cancer tests for assessing human risk at low doses need reexamination. No diet can be free of natural chemicals that are rodent carcinogens.53
[page]"About 50 percent of all chemicals – natural or synthetic – that have been tested in standard, high-dose animal cancer tests are rodent carcinogens."
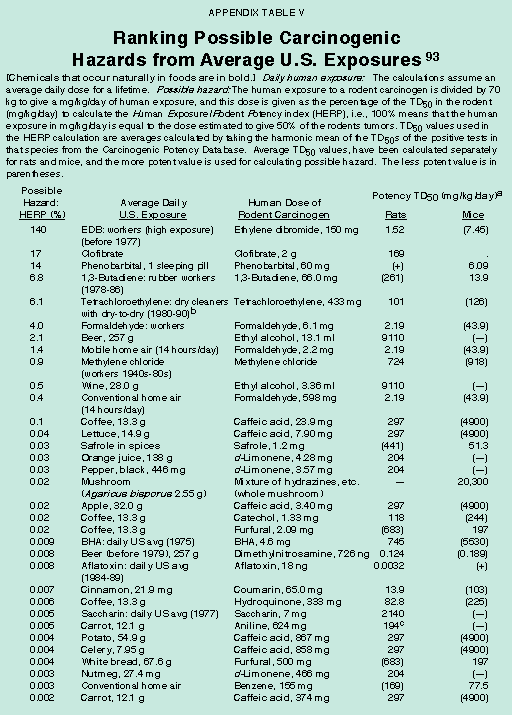
About 50 percent of all chemicals – whether natural or synthetic – that have been tested in standard, high-dose, animal cancer tests are rodent carcinogens.54 [See Appendix Table IV.] What are the explanations for this high percentage? In standard cancer tests rodents are given chronic, near-toxic doses – the maximum tolerated dose (MTD). Evidence is accumulating that cell division caused by the high dose itself, rather than the chemical per se, can contribute to cancer in these tests. High doses can cause chronic wounding of tissues, cell death and consequent chronic cell division of neighboring cells, which is a risk factor for cancer.55 Each time a cell divides, the probability increases that a mutation will occur, thereby increasing the risk for cancer. At the low levels to which humans are usually exposed, such increased cell division does not occur. In addition, tissues injured by high doses of chemicals have an inflammatory immune response involving activation of white cells in response to cell death.56 Activated white cells release mutagenic oxidants (including peroxynitrite, hypochlorite, and hydrogen peroxide). Therefore, the very low levels of chemicals to which humans are exposed through water pollution or synthetic pesticide residues may pose no or minimal cancer risks.
The authors have discussed in another paper57 the argument that the high positivity rate is due to selecting more suspicious chemicals to test, which is a likely bias since cancer testing is both expensive and time-consuming, and it is prudent to test suspicious compounds. One argument against selection bias is the high positivity rate for drugs [see Appendix Table IV] because drug development tends to select chemicals that are not mutagens or expected carcinogens. A second argument against selection bias is that knowledge to predict carcinogenicity in rodent tests is highly imperfect, even now after decades of testing results have become available on which to base prediction. For example, a prospective prediction exercise was conducted by several experts in 1990 in advance of the two-year NTP bioassays. There was wide disagreement among them on which chemicals would be carcinogenic when tested, and accuracy varied, thus indicating that predictive knowledge is highly uncertain.58
"It seems likely that a high proportion of all chemicals, whether synthetic or natural, might be ‘carcinogens' if administered in high enough doses."
It seems likely that a high proportion of all chemicals, whether synthetic or natural, might be "carcinogens" if administered in the standard rodent bioassay at the maximum tolerated dose, primarily due to the effects of high doses on cell division and DNA damage.59 Without additional data on how a chemical causes cancer, the interpretation of a positive result in a rodent bioassay is highly uncertain. The induction of cancer could be the result of the high doses tested.
In regulatory policy, the "virtually safe dose" (VSD), corresponding to a maximum, hypothetical risk of one cancer in a million, is estimated from bioassay results using a linear model, which assumes that cancer causation is directly proportional to dose and that there are no unique effects of high doses. To the extent that carcinogenicity in rodent bioassays is due to the effects of high doses for the non-mutagens, and a synergistic effect of cell division at high doses with DNA damage for the mutagens, then this model is inappropriate. The EPA has recently proposed guidelines that permit the use of non-linear approaches to low dose extrapolation if warranted by mechanistic data and a possible threshold of dose below which effects will not occur.60
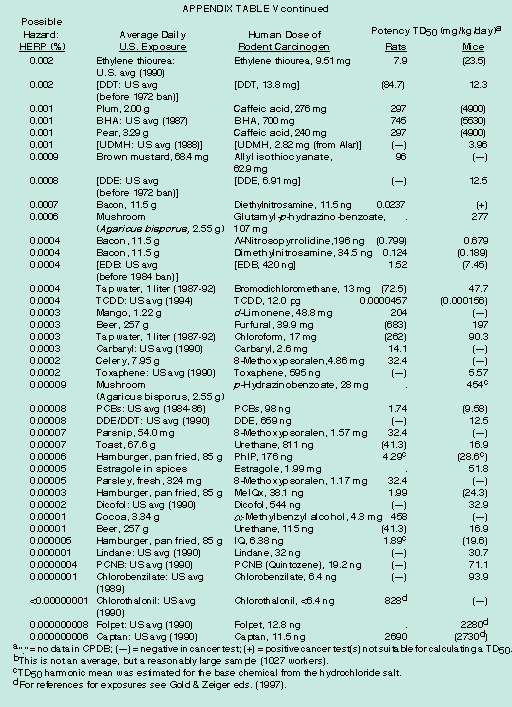
[page]Gaining a broad perspective about the vast number of chemicals to which humans are exposed can be helpful when setting research and regulatory priorities.61 Rodent cancer tests provide little information about how a chemical causes cancer or about low-dose risk. The assumption that synthetic chemicals are hazardous has led to a bias in testing, such that synthetic chemicals account for 77 percent (432 of 559) of the chemicals tested chronically in both rats and mice. [See Appendix Table IV.] The natural world of chemicals has never been tested systematically. One reasonable strategy is to use a rough index to compare and rank possible carcinogenic hazards from a wide variety of chemical exposures at levels that humans typically receive, and then to focus on those that rank highest.62 Ranking is a critical first step that can help to set priorities for selecting chemicals for long term cancer tests, studies on mechanism, epidemiological research and regulatory policy. Although one cannot say whether the ranked chemical exposures are likely to be of major or minor importance in human cancer, it is not prudent to focus attention on the possible hazards at the bottom of a ranking if, using the same methodology to identify hazard, there are numerous common human exposures with much greater possible hazards. Our analyses are based on the HERP index (Human Exposure/Rodent Potency), which indicates what percentage of the rodent carcinogenic potency (TD50 in mg/kg/day, i.e. dose to give 50 percent of test animals tumors) a human receives from a given daily lifetime exposure (mg/kg/day).63 [See Appendix Table Va and Vb.] A ranking based on standard regulatory risk assessment would be similar.
"Our results call for a reevaluation of the utility of animal cancer tests in protecting the public against minor hypothetical risks."
Overall, our analyses have shown that HERP values for some historically high exposures in the workplace (e.g., butadiene and tetrachloroethylene) and some pharmaceuticals (e.g., clofibrate) rank high, and that there is an enormous background of naturally occurring rodent carcinogens in typical portions of common foods that cast doubt on the relative importance of low-dose exposures to residues of synthetic chemicals such as pesticides.64 A committee of the National Research Council/National Academy of Sciences recently reached similar conclusions about natural vs. synthetic chemicals in the diet, and called for further research on natural chemicals.65
The possible carcinogenic hazards from synthetic pesticides (at average exposures) are minimal compared to the background of nature's pesticides, though neither may be a hazard at the low doses consumed. [See Appendix Table Va and Vb.] Appendix Table V also indicates that many ordinary foods would not pass the regulatory criteria used for synthetic chemicals. For many natural chemicals the HERP values are in the top half of the table, even though natural chemicals are markedly underrepresented because so few have been tested in rodent bioassays. Caution is necessary in drawing conclusions from the occurrence in the diet of natural chemicals that are rodent carcinogens. It is not argued here that these dietary exposures are necessarily of much relevance to human cancer. Our results call for a reevaluation of the utility of animal cancer tests in protecting the public against minor hypothetical risks.
[page]It is often assumed that because natural chemicals are part of human evolutionary history, whereas synthetic chemicals are recent, the mechanisms that have evolved in animals to cope with the toxicity of natural chemicals will fail to protect against synthetic chemicals. This assumption is flawed for several reasons.66
a) Humans have many natural defenses that buffer against normal exposures to toxins,67 and these are usually general, rather than tailored for each specific chemical. Thus they work against both natural and synthetic chemicals. Examples of general defenses include the continuous shedding of cells exposed to toxins – the surface layers of the mouth, esophagus, stomach, intestine, colon, skin and lungs are discarded every few days; DNA repair enzymes, which repair DNA that was damaged from many different sources; and detoxification enzymes of the liver and other organs which generally target classes of toxins rather than individual toxins. That human defenses are usually general, rather than specific for each chemical, makes good evolutionary sense. The reason that predators of plants evolved general defenses is presumably to be prepared to counter a diverse and ever-changing array of plant toxins in an evolving world; if a herbivore had defenses against only a set of specific toxins, it would be at a great disadvantage in obtaining new food when favored foods became scarce or evolved new toxins.
b) Various natural toxins, which have been present throughout vertebrate evolutionary history, nevertheless cause cancer in vertebrates.68 Mold toxins, such as aflatoxin, have been shown to cause cancer in rodents and other species including humans [see Appendix Table IV.] Many of the common elements are carcinogenic to humans at high doses (e.g., salts of cadmium, beryllium, nickel, chromium and arsenic) despite their presence throughout evolution. Furthermore, epidemiological studies from various parts of the world show that certain natural chemicals in food may be carcinogenic risks to humans; for example, the chewing of betel nuts with tobacco has been correlated with oral cancer.
"Natural selection works far too slowly for humans to have evolved specific resistance to the toxins in many common foods."
c) Humans have not had time to evolve a "toxic harmony" with all of their dietary plants. The human diet has changed markedly in the last few thousand years. Indeed, very few of the plants that humans eat today (e.g., coffee, cocoa, tea, potatoes, tomatoes, corn, avocados, mangoes, olives and kiwi fruit) would have been present in a hunter-gatherer's diet. Natural selection works far too slowly for humans to have evolved specific resistance to the food toxins in these newly introduced plants.
"There is no convincing epidemiological evidence that the levels of DDT normally found in the environment are likely to be a significant contributor to cancer."
d) DDT is often viewed as the typically dangerous synthetic pesticide because it concentrates in the tissues and persists for years, being slowly released into the bloodstream. DDT, the first synthetic pesticide, eradicated malaria from many parts of the world, including the U.S. It was effective against many vectors of disease such as mosquitoes, tsetse flies, lice, ticks and fleas. DDT was also lethal to many crop pests, and significantly increased the supply and lowered the cost of food, making fresh nutritious foods more accessible to poor people. DDT was also of low toxicity to humans. A 1970 National Academy of Sciences report concluded: "In little more than two decades DDT has prevented 500 million deaths due to malaria, that would otherwise have been inevitable."69 There is no convincing epidemiological evidence, nor is there much toxicological plausibility, that the levels of DDT normally found in the environment are likely to be a significant contributor to cancer. DDT was unusual with respect to bioconcentration, and because of its chlorine substituents it takes longer to degrade in nature than most chemicals; however, these are properties of relatively few synthetic chemicals. In addition, many thousands of chlorinated chemicals are produced in nature70 and natural pesticides also can bioconcentrate if they are fat soluble. Potatoes, for example, naturally contain the fat soluble neurotoxins solanine and chaconine, which can be detected in the bloodstream of all potato eaters. High levels of these potato neurotoxins have been shown to cause birth defects in rodents.71
e) Since no plot of land is immune to attack by insects, plants need chemical defenses – either natural or synthetic – to survive pest attack. Thus, there is a trade-off between naturally occurring pesticides and synthetic pesticides. One consequence of disproportionate concern about synthetic pesticide residues is that some plant breeders develop plants to be more insect-resistant by making them higher in natural toxins. A recent case illustrates the potential hazards of this approach to pest control: When a major grower introduced a new variety of highly insect-resistant celery into commerce, people who handled the celery developed rashes when they were subsequently exposed to sunlight. Some detective work found that the pest-resistant celery contained 6,200 parts per billion (ppb) of carcinogenic (and mutagenic) psoralens instead of the 800 ppb present in common celery.72
[page]"Even if sperm counts were declining, there are many more likely causes, such as smoking and diet."
Synthetic hormone mimics have become an environmental issue. Hormonal factors are important in cancer, as mentioned in Misconception #2. The 1996 book Our Stolen Future73 claims that traces of synthetic chemicals, such as pesticides with weak hormonal activity, may contribute to cancer and reduce sperm counts. The book ignores the fact that our normal diet contains natural chemicals that have estrogenic activity millions of times higher than that due to the traces of synthetic estrogenic chemicals74 and that lifestyle factors can markedly change the levels of endogenous hormones [see Misconception #2]. The low levels of exposure to residues of industrial chemicals in humans are toxicologically implausible as a significant cause of cancer or of reproductive abnormalities, especially when compared to the natural background.75 In addition, it has not been shown convincingly that sperm counts are declining,76 and even if they were, there are many more likely causes, such as smoking and diet.
[page]"One estimate is that the U.S. could prevent 60,000 deaths per year by redirecting current resources to more cost-effective programs."
Since there is no risk-free world and resources are limited, society must set priorities based on cost-effectiveness in order to save the most lives.77 The EPA projected in 1991 that the cost to society of environmental regulations in 1997 would be about $140 billion per year (about 2.6 percent of gross national product).78 Most of this cost is to the private sector. Several economic analyses have concluded that current expenditures are not cost-effective; that is, resources are not being utilized so as to save the most lives per dollar. One estimate is that the U.S. could prevent 60,000 deaths per year by redirecting the same dollar resources to more cost-effective programs.79 For example, the median toxin control program costs 146 times more per year of life saved than the median medical intervention.80 This difference is likely to be greater, because cancer risk estimates for toxin control programs are worst-case, hypothetical estimates, and the true risks at low dose are often likely to be zero.81 [See Misconception #6.] Some economists have argued that costly regulations intended to save lives may actually lead to increased deaths,82 in part because they divert resources from important health risks and in part because higher incomes are associated with lower mortality.83 Rules on air and water pollution are necessary (e.g., it was a public health advance to phase lead out of gasoline) and clearly, cancer prevention is not the only reason for regulations. However, worst-case assumptions in risk assessment represent a policy decision, not a scientific one, and they confuse attempts to allocate money effectively for risk abatement.
Regulatory efforts to reduce low-level human exposures to synthetic chemicals because they are rodent carcinogens are expensive because they aim to eliminate minuscule concentrations that now can be measured with improved techniques. These efforts are distractions from the major task of improving public health through increasing scientific understanding about how to prevent cancer (e.g., the role of diet), increasing public understanding of how lifestyle influences health and improving our ability to help individuals alter lifestyle.
Why has the government focused on minor hypothetical risks at huge cost? A recent article in The Economist84 had a fairly harsh judgment: "Predictions of ecological doom, including recent ones have such a terrible track record that people should take them with pinches of salt instead of lapping them up with relish. For reasons of their own, pressure groups, journalists and fameseekers will no doubt continue to peddle ecological catastrophes at an undiminishing speed…. Environmentalists are quick to accuse their opponents in business of having vested interests. But their own incomes, their fame and their very existence can depend on supporting the most alarming versions of every environmental scare. 'The whole aim of practical politics' said H.L. Mencken, 'is to keep the populace alarmed – and hence clamorous to be led to safety – by menacing it with a series of hobgoblins, all of them imaginary.' Mencken's forecast, at least, appears to have been correct."
Dr. Aaron Wildavsky discusses worst-case risk assessment in his book But Is It True: A Citizen's Guide to Environmental Health and Safety Issues.85 "We should be guided by the probability and extent of harm, not by its mere possibility. The search for possibilities is endless and it trivializes the subject. There is bound to be great diversion of resources without reducing substantial sources of harm. Consternation is created but health is not enhanced…. Weak causes are likely to have weak effects. Our search should be for strong causes with palpable effects, like cigarette smoking. They are easier to find and their effects are much more important to control…. The past necessity of proving harm has been replaced by a reversal of causality: now the individuals and businesses must prove that they will do no harm. My objection to this…is profound: our liberties are curbed and our health is harmed."
Acknowledgments. This paper is modified from testimony March 6, 1997 for the U.S. Senate Hearing on Environmental Risk Factors for Cancer and FASEB J. Vol. 11, 1997. This work was supported by the National Cancer Institute Outstanding Investigator Grant CA39910 to B.N.A., the Director, Office of Energy Research, Office of Health and Environmental Research of the U.S. Department of Energy under Contract DE-AC03-76SF00098 to L.S.G., and the National Institute of Environmental Health Sciences Center Grant ESO1896.
NOTE: Nothing written here should be construed as necessarily reflecting the views of the National Center for Policy Analysis or as an attempt to aid or hinder the passage of any bill before Congress.
[page]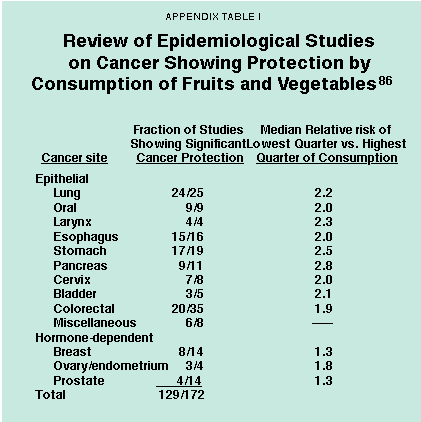



-
Ries, L. A. G., Kosary, C. L., Hankey, B. F., Miller, B. A., Harras, A. & Edwards, B. K. (1997) SEER Cancer Statistics Review, 1973-1994 (National Cancer Institute, Bethesda, Md.).
- Devesa, S., Blot, W. J., Stone, B. J., Miller, B. A., Tarone, R. E. & Fraumeni, F. J., Jr. (1995) Journal of the National Cancer Institute 87, 175-82; Doll, R. & Peto, R. (1981) Journal of the National Cancer Institute 66, 1191-1308; and Ames et al. (1995).
- Bailar, I., J C & Gornik, H. L. (1997) New England Journal of Medicine 336, 1569-1574.
- Gold, L. S., Slone, T. H., Stern, B. R., Manley, N. B. & Ames, B. N. (1992) Science 258, 261-65; and Ames, B. N., Gold, L. S. & Willett, W. C. (1995) Proceedings of the National Academy of Sciences USA 92, 5258-65.
- Gold et al. (1992); National Research Council (1996) Carcinogens and Anticarcinogens in the Human Diet: A Comparison of Naturally Occurring and Synthetic Substances (National Academy Press, Washington, D.C.); and Gold, L.S. & Zeiger, E. (1997) Handbook of Carcinogenic Potency and Genotoxicity Databases (CRC Press, Baco Raton, FL).
- Gough, M. (1990) Risk Analysis 10, 1-6.
- Ames et al. (1995).
- Gold, L. S., Garfinkel, G. B. & Slone, T. H. (1994) in Chemical Risk Assessment and Occupational Health, Current Applications, Limitations, and Future Prospects, eds. Smith, C. M., Christiani, D. C. & Kelsey, K. T. (Greenwood Publishing Group, Westport, Conn.), pp. 91-103.
- Ames, B. N., Shigenaga, M. K. & Hagen, T. M. (1993) Proceedings of the National Academy of Sciences USA 90, 7915-22.
- Ibid.
- Hagen, T. M., Yowe, D. L., Bartholomew, J. C., Wehr, C. M., Do, K. L., Park, J.-Y. & Ames, B. N. (1997) Proceedings of the National Academy of Sciences USA 94, 3064-69.
- Helbock, H. J., Beckman, K. B., Shigenaga, M. K., Walter, P., Woodall, A. A., Yeo, H. C. & Ames, B. N. (1997) Proceedings of the National Academy of Sciences, 95, 288-93.
- Rice-Evans, C., Sampson, J., Bramley, P. & Holloway, D. (1997) Free Radical Research 26, 381-98.
- Devesa et al. (1995); and Peto, R., Lopez, A. D., Boreham, J., Thun, M. & Heath, C., Jr. (1994) Mortality from Smoking in Developed Countries 1950-2000 (Oxford University Press, Oxford, England).
- Fraga, C. G., Motchnik, P. A., Shigenaga, M. K., Helbock, H. J., Jacob, R. A. & Ames, B. N. (1991) Proceedings of the National Academy of Sciences USA 88, 11003-06; Ames, B. N., Motchnik, P. A., Fraga, C. G., Shigenaga, M. K. & Hagen, T. M. (1994) in Male-Mediated Developmental Toxicity, eds. Mattison, D. R. & Olshan, A. (Plenum Publishing Corporation, New York), pp. 243-59; and Fraga, C. G., Motchnik, P. A., Wyrobek, A. J., Rempel, D. M. & Ames, B. N. (1996) Mutation Research 351, 199-203.
- Fraga et al. (1996).
- Wyrobek, A. J., Rubes, J., Cassel, M., Moore, D., Perrault, S., Slott, V., Evenson, D., Zudova, Z., Borkovec, L., Selevan, S. & Lowe, X. (1995) American Journal of Human Genetics 57, 737.
- Fraga et al. (1991); Ames et al. (1994) in Mattison & Olshan, eds.; and Woodall, A. A. & Ames, B. N. (1997) in Preventative Nutrition: The Comprehensive Guide for Health Professionals, eds. Bendich, A. Decklbaum, R.J. (Humana Press, Totowa, NJ), pp. 373-385.
- Ji, B.-T., Shu, X.-O., Linet, M. S., Zheng, W., Wacholder, S., Gao, Y.-T., Ying, D.-M. & Jin, F. (1997) Journal of the National Cancer Institute 89, 238-244.
- Doll, R. & Peto, R. (1981).
- Ames et al. (1995); Hart, R., Keenan, K., Turturro, A., Abdo, K., Leakey, J. & Lyn-Cook, B. (1995) Fundamentals of Applied Toxicology, 184-95; and Turturro, A., Duffy, P., Hart, R. & Allaben, W. (1996) Toxicological Pathology 24, 769-75.
- Ames et al. (1995); Christen, S., Hagen, T. M., Shigenaga, M. K. & Ames, B. N. (1998) in Microbes and Malignancy: Infection as a Cause of Cancer, eds. Parsonnet, J. & Horning, S. (Oxford University Press, Oxford), pp. in press.
- Shacter, E., Beecham, E. J., Covey, J. M., Kohn, K. W. & Potter, M. (1988) Carcinogenesis 9, 2297-2304; and Yamashina, K., Miller, B. E. & Heppner, G. H. (1986) Cancer Research 46, 2396-2401.
- Pisani, P., Parkin, D. M., Muñoz, N. & Ferlay, J. (1997) Cancer Epidemiology Biomarkers & Prevention 6, 387-400.
- Henderson, B. E., Ross, R. K. & Pike, M. C. (1991) Science 254, 1131-38; and Feigelson, H. S. & Henderson, B. E. (1996) Carcinogenesis 17, 2279-84.
- Ibid.; and Hunter, D. J. & Willett, W. C. (1993) Epidemiologic Reviews 15, 110-132.
- Ames et al. (1995); Ames et al. (1993) Proceedings of the National Academy of Sciences.
- Block, G., Patterson, B. & Subar, A. (1992) Nutrition and Cancer 18, 1-29; Steinmetz, K. A. & Potter, J. D. (1996) Journal of the American Dietary Association 96, 1027-39; and Hill, M. J., Giacosa, A. & Caygill, C. P. J. (1994) Epidemiology of Diet and Cancer (Ellis Horwood Limited, West Sussex, England).
- Krebs-Smith, S.M., Cook, A., Subar, A. F., Cleveland, L., Friday, J., and Kahle, L. L. (1996) Archives of Pediatrics and Adolescent Medicine 150, 81-6; and Krebs-Smith, S. M., Cook, A., Subar, A. F., Cleveland, L., and Friday, J. (1995) American Journal of Public Health 85, 1623-29.
- A National Cancer Institute Graphic (1996) Journal of the National Cancer Institute 88, 1314.
- Steinmetz et al. (1996); Hill et al. (1994); Block, G. (1992) Nutrition Reviews 50, 207-213.
- Blount, B. C., Mack, M. M., Wehr, C., MacGregor, J., Hiatt, R., Wang, G., Wickramasinghe, S. N., Everson, R. B. & Ames, B. N. (1997) Proceedings of the National Academy of Sciences USA 94, 3290-95.
- Senti, F. R. & Pilch, S. M. (1985) Journal of Nutrition 115, 1398-402.
- Blount et al. (1997).
- Bailey, L. B., Wagner, P. A., Christakis, G. J., Araujo, P. E., Appledorf, H., Davis, C. G., Masteryanni, J. & Dinning, J. S. (1979) American Journal of Clinical Nutrition 32, 2346-53.
- Bailey, L. B., Wagner, P. A., Christakis, G. J., Davis, C. G., Appledorf, H., Araujo, P. E., Dorsey, E. & Dinning, J. S. (1982) American Journal of Clinical Nutrition 35, 1023-32.
- Blount et al. (1997).
- Ibid.
- Wallock, L., Woodall, A., Jacob, R. & Ames, B. (1997) Nutritional status and positive relation of plasma folate to fertility indices in nonsmoking men (The FASEB J., New Orleans, LA), Vol. 11, p. A184 (Abstracts).
- Boushey, C. J., Beresford, S. A., Omenn, G. S. & Motulsky, A. G. (1995) Journal of the American Medical Association 274, 1049-57.
- Ames, et al. (1995); Block et al. (1992); and Subar, A. F., Block, G. & James, L. D. (1989) American Journal of Clinical Nutrition 50, 508-16.
- Herbert, V. (1996) in Present Knowledge in Nutrition, eds. Ziegler, E. E. & Filer, L. J. (ILSI Press, Washington, D.C.), pp. 191-205.
- Wickramasinghe, S. N. & Fida, S. (1994) Blood 83, 1656-61.
- Herbert, V. (1996).
- Zhang, J. Z., Henning, S. M. & Swendseid, M. E. (1993) Journal of Nutrition 123, 1349-55.
- Jacobson, E. L. (1993) Journal of American College of Nutrition 12, 412-6.
- Ames, B. N. (1998) Manuscript in preparation.
- Ames, B. N., Profet, M. & Gold, L. S. (1990) Proceedings of the National Academy of Sciences USA 87, 7777-81 and 7782-86; and Gold, L. S., Slone, T. H. & Ames, B. N. (1997) in Food Chemical Risk Analysis, ed. Tennant, D. (Chapman & Hall Ltd, London), pp. 267-295.
- Ibid.
- Gold et al. (1997).
- Ames et al. (1990) 7777-81; Gold et al. (1997) in Tennant, ed.
- Gold et al. (1992).
- Gold et al. (1997) in Tennant, ed.
- Ames, B. N., Gold, L. S. & Shigenaga, M. K. (1996) Risk Analysis 16, 613-17; and Gold, L. S., Slone, T. H. & Ames, B. N. (1997) in Handbook of Carcinogenic Potency and Genotoxicity Database, eds. Gold, L. S. & Zeiger, E. (CRC Press, Boca Raton, FL), pp. 661-85.
- Ames et al. (1996).
- Laskin, D. L. & Pendino, K. J. (1995) Annual Review of Pharmacology and Toxicology 35, 655-77; Wei, H. & Frenkel, K. (1993) Carcinogenesis 14, 1195-1201; Wei, L., Wei, H. & Frenkel, K. (1993) Carcinogenesis 14, 841-47; Laskin, D. L., Robertson, F. M., Pilaro, A. M. & Laskin, J. D. (1988) Hepatology 8, 1051-55; Czaja, M. J., Xu, J., Ju, Y., Alt, E. & Schmiedeberg, P. (1994) Hepatology 19, 1282-89; Adachi, Y., Moore, L. E., Bradford, B. U., Gao, W. & Thurman, R. G. (1995) Gastroenterology 108, 218-24; and Gunawardhana, L., Mobley, S. A. & Sipes, I. G. (1993) Toxicology and Applied Pharmacology 119, 205-213.
- Gold, L. S., Slone, T. H. & Ames, B. N. (1998) Drug Metabolism Reviews, 30, 361-405.
- Omenn, G. S., Stuebbe, S. & Lave, L. B. (1995) Molecular Carcinogenesis 14, 37-45.
- Butterworth, B., Conolly, R. & Morgan, K. (1995) Cancer Letters 93, 129-46; Ames, B. N., Shigenaga, M. K. & Gold, L. S. (1993) Environmental Health Perspectives 101 (Suppl 5), 35-44; and Ames, B. N. & Gold, L. S. (1990) Proceedings of the National Academy of Sciences USA 87, 7772-76.
- Environmental Protection Agency (1996), Proposed guidelines for carcinogenic risk assessment, Vol. 61, 17960-18011.
- Gold et al. (1992); Ames et al. (1990) 7782-86; Gold et al. (1997) in Tennant, ed; and Ames, B. N., Magaw, R. & Gold, L. S. (1987) Science 236, 271-80.
- Gold et al. (1992); and Gold et al. (1997) in Gold and Zeiger, eds.
- Gold, L. S. & Zeiger, E., eds. (1997).
- Gold et al. (1992); Gold et al. (1994); and Gold et al. (1997) in Gold and Zeiger, eds.
- National Research Council (1996).
- Ames et al. (1990) 7782-86; and Ames et al. (1996).
- Ames et al. (1990) 7782-86.
- Ibid.; and Gold et al. (1997) in Gold and Zeiger, eds.
- National Academy of Sciences, U. S. A. (1970), The Life Sciences: Recent Progress and Application to Human Affairs, the World of Biological Research, Requirement for the Future (Committee on Research in the Life Sciences, Washington, D.C.).
- Gribble, G. W. (1996), Pure & Applied Chemistry 68, 1699-1712.
- Ames et al. (1990) 7782-86.
- Ibid.
- Colburn, T., Dumanoski, D. & Meyers, J. P. (1996) Our Stolen Future: Are We Threatening Our Fertility, Intelligence, and Survival?: A Scientific Detective Story (Dutton, New York).
- Safe, S. H. (1994) Environ. Sci. Pollution Res. 1, 29-33; and Safe, S. H. (1995) Environmental Health Perspectives 103, 346-51.
- Safe (1994); Safe (1995); Safe, S. H. (1997) Environmental Health Perspectives 105, (Suppl 3): 675-78; and Reinli, K. & Block, G. (1996) Nutrition and Cancer 26, 1996.
- Kolata, G. (1996) Measuring men up, sperm by sperm, The New York Times, E4(N), E4(L), (col.1), May 4.
- Hahn, R. W., ed. Risks, Costs, and Lives Saved: Getting Better Results from Regulation (Oxford University Press and AEI Press, New York and Washington, DC); and Graham, J. & Wiener, J., eds. Risk versus Risk: Tradeoffs in Protecting Health and the Environment (Harvard University Press, Cambridge, Massachusetts).
- U.S. Environmental Protection Agency (1991) Environmental Investments: The Cost of a Clean Environment (Office of the Administrator, Washington, D. C.).
- Tengs et al. (1995) Risk Analysis 15, 369-89.
- Ibid.
- Gold et al. (1992); Ames et al. (1995); and Gold et al. (1997) in Gold and Zeiger, eds.
- Keeney, R. L. (1990) Risk Analysis 10, 147-59.
- Wildavsky, A. (1988) Searching for Safety (Transaction Press, New Brunswick, N.J.); Wildavsky, A. B. (1995) But is it True?: A Citizen's Guide to Environmental Health and Safety (Harvard University Press, Cambridge, MA); and Viscusi, W. K. (1992) Fatal Trade-offs (Oxford University Press, Oxford, England).
- (1997) Environmental Scares: Plenty of Gloom, The Economist, pp. 19-21.
- Wildavsky, A.B. (1995) But is it true?: A Citizen's Guide to Environmental Health and Safety, (Harvard University Press, Cambridge, MA).
- Block et al. (1992).
- Gold et al. (1997) in Tennant, ed.
- Gold et al. (1997) in Gold & Zeiger, eds.
- Ibid.
- Innes, J. R. M., Ulland, B. M., Valerio, M. G., Petrucelli, L., Fishbein. L., Hart, E. R., Pallota, A. J., Bates R. R., Falk, H. L., Gart, J. J., Klein, M., Mitchell, I. & Peters, J. (1969) Journal of the National Cancer Institute 42, 1101-14.
- Davies, T. S. & Monro, A. (1995) Journal of the American College of Toxicology 14, 90-107.
- Contrera, J., Jacobs, A. & DeGeorge, J. (1997) Regulatory Toxicology and Pharmacology 25, 130-45.
- Gold et al. (1997) in Gold & Zeiger, eds.
Dr. Bruce N. Ames is a Professor of Biochemistry and Molecular Biology and Director of the National Institute of Environmental Health Sciences Center, University of California, Berkeley. He is a member of the National Academy of Sciences and was on their Commission on Life Sciences. He was a member of the National Cancer Advisory Board of the National Cancer Institute (1976-82). His many awards include: the General Motors Cancer Research Foundation Prize (1983), the Tyler Prize for environmental achievement (1985), the Gold Medal Award of the American Institute of Chemists (1991), the Glenn Foundation Award of the Gerontological Society of America (1992), the Lovelace Institutes Award for Excellence in Environmental Health Research (1995), the Honda Foundation Prize for Ecotoxicology (1996) and the Japan Prize (1997). His 400+ publications have resulted in his being the 23rd most-cited scientist (in all fields) (1973-84). BNAmes@UCLink4.Berkeley.edu
Dr. Lois Swirsky Gold is Director of the Carcinogenic Potency Project at the National Institute of Environmental Health Sciences Center, University of California, Berkeley, and a Senior Scientist at the Berkeley National Laboratory 94720. She has published 85 papers on analyses of animal cancer tests and implications for cancer prevention, interspecies extrapolation and regulatory policy. The Carcinogenic Potency Database (CPDB), published as a CRC handbook, analyzes results of 5000 chronic, long-term cancer tests on 1300 chemicals. Dr. Gold has served on the Panel of Expert Reviewers for the National Toxicology Program, the Boards of the Harvard Center for Risk Analysis and the Annapolis Center, and was a member of the Harvard Risk Management Group. Lois@potency.Berkeley.edu